Mutation, Selection, and Drift at a Single Codon
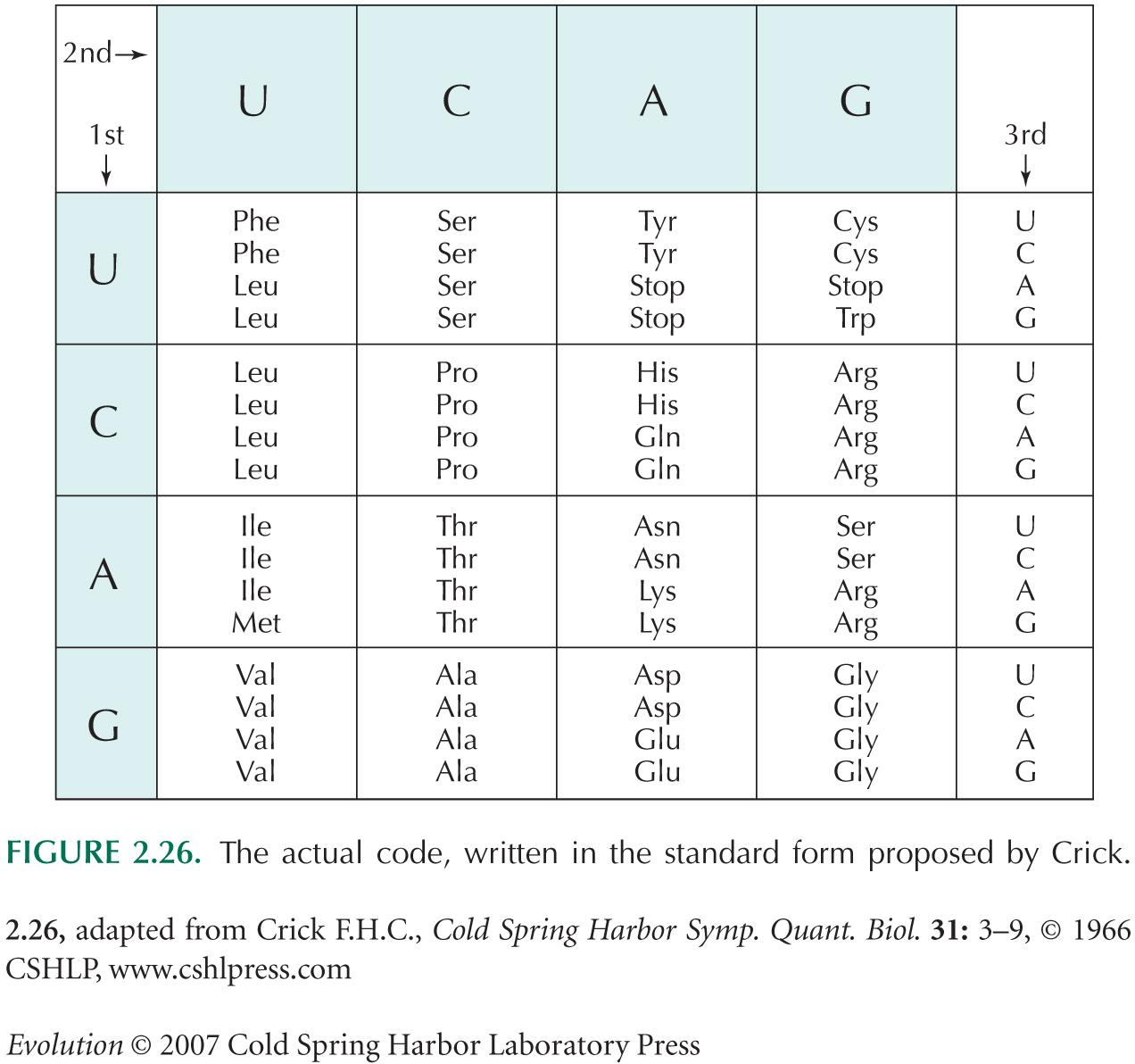
Suppose that at a particular position in a protein, one amino acid is optimal. Mutations at the third nucleotide position within the codon that codes for this amino acid usually leave the amino acid unchanged. However, mutations at either of the other two positions within the codon are likely to result in a different amino acid (look at a copy of the genetic code to see why this is the case; e.g., see Fig. 2.26). The rate at which the optimal amino acid is replaced by a less-fit alternative is the product of the rate of mutations that alter the protein sequence (~2µ, because there are two positions likely to alter the amino acid sequence; µ is the mutation rate per nucleotide per generation); the number of genes in the population (2N); and the chance that any one mutation will fix, ~2s(Ne/N)e–4Nes, where s is the fitness loss in the heterozygote. Overall, then, the rate of loss of this optimal amino acid is approximately 8Nes µ e–4Nes. Once it is lost, only one particular mutation can precisely restore the same DNA sequence. (In some cases, a different DNA sequence could restore the same amino acid sequence, but it is a fair approximation to ignore this possibility.) The rate of reverse mutations is thus µ/3, because only one of the three possible changes at a site will restore the original sequence. By the same reasoning as before, there are 2N genes in the whole population, and the chance that any one reverse mutation will fix is approximately 2s(Ne/N); the net rate at which the original allele is recovered is therefore (2Nesµ/3).
The ratio between the chances that a site is occupied by a suboptimal amino acid versus the optimal amino acid is therefore the ratio of the two rates, 6 e–4Nes. This depends just on the ratio of rates of deleterious versus reverse mutations (2µ vs. µ/3) and on the factor e–4Nes, which gives the relative effectiveness of selection versus drift. If Nes is small, then single-error variants become common, and further mutations may take the sequence even further from the optimum. Function is then likely to collapse completely under the combined pressure of mutation and random drift. (See Fig. WN18.2.)
|



{kind=link}