Chapter 26 Notes
Current Issues in Human Evolution
The Genetic Basis of Disease
Why Study Disease Genetics?
The incidence of cystic fibrosis is discussed by Vogel and Motulsky (1997, pp. 111, 290).
Genetic factors that predispose to diabetes, including PPARg, are reviewed by O’Rahilly et al. (2005).
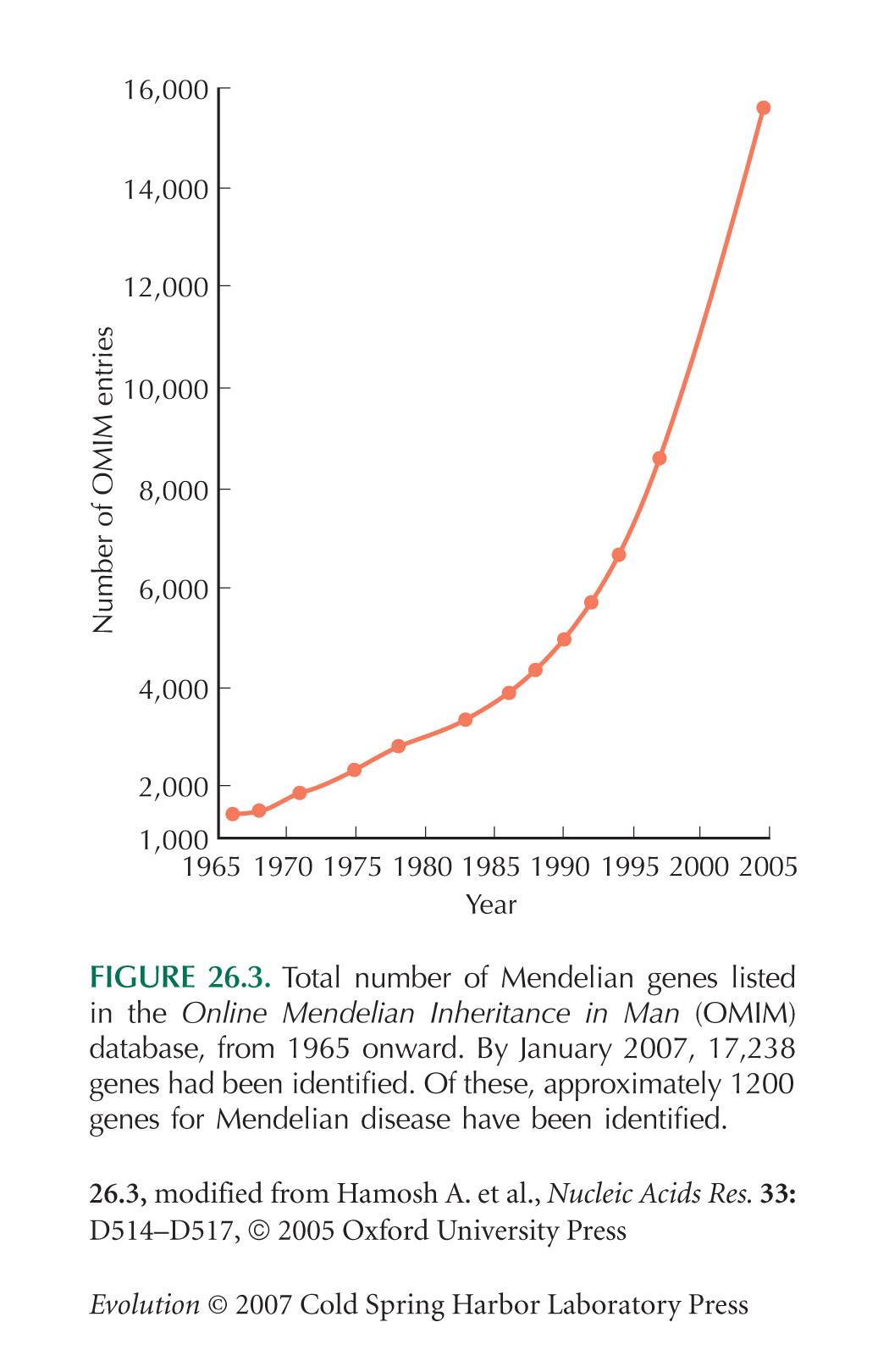
The Online Mendelian Inheritance in Man (OMIM) database catalogs genes known to cause human disease.
The Genes Responsible for Mendelian Disease Can Be Mapped
Morton (1995) gives a history of statistical methods for mapping Mendelian loci in humans; see also Terwilliger and Ott (1994) and Ott (1999). Bell and Haldane (1937) gave the first evidence for linkage in humans, between the X-linked genes for color blindness and hemophilia. Demonstrating linkage between autosomal genes is much harder. Bernstein (1931) set out a method for detecting linkage between autosomal loci in humans (see Crow 1993). This was later superseded by methods based on likelihood, introduced by Haldane and Smith (1947). Because of the lack of genetic markers in humans, the first autosomal linkages were not actually discovered until the 1950s (see Morton 1995).
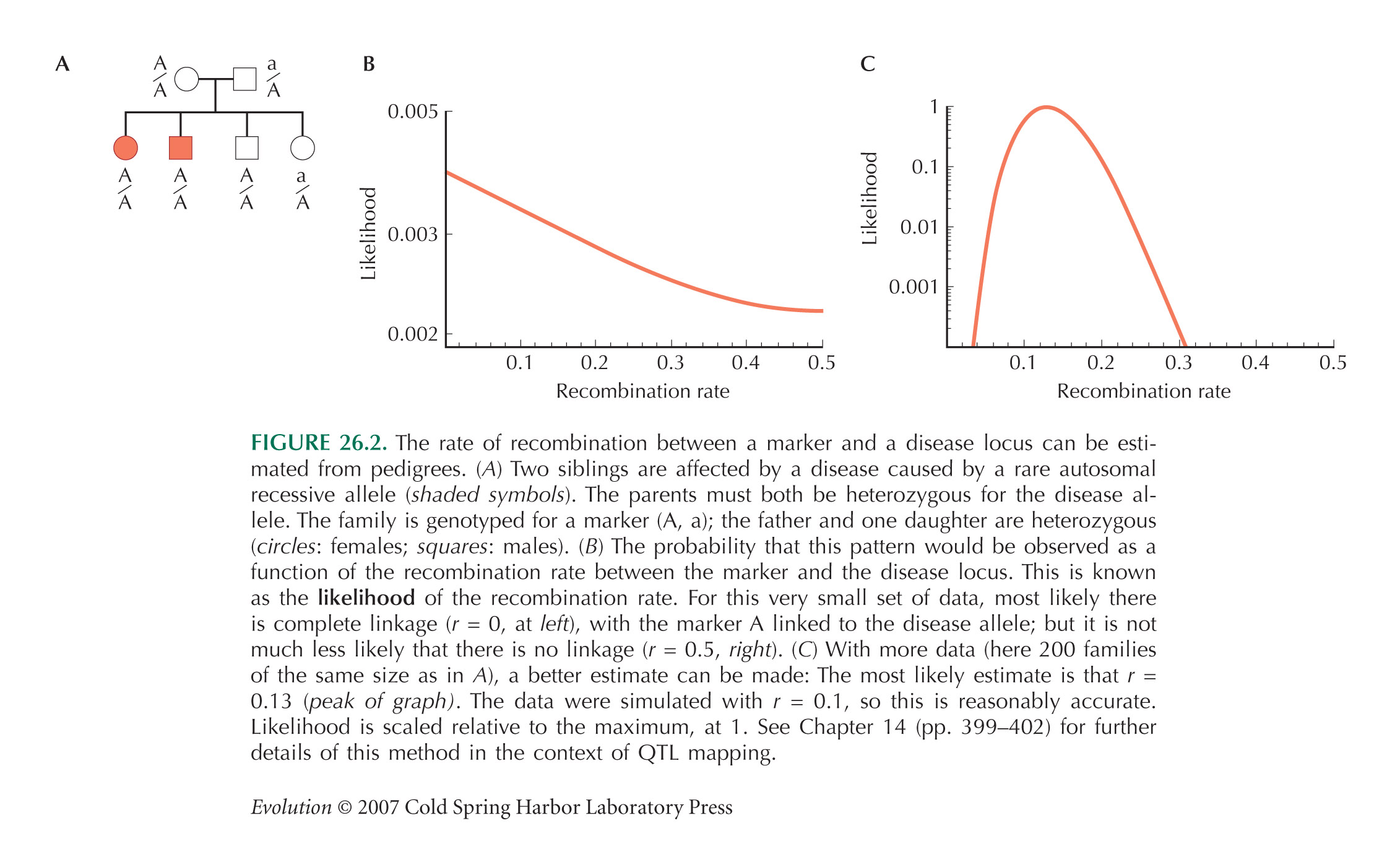
Figure 26.2A shows a pedigree with four offspring, two of which are affected by an autosomal recessive disease. Both parents must be heterozygous for the disease allele (D/d, say). The two affected siblings are both homozygous A/A for a marker allele, whereas of the two unaffected siblings, one is A/a and the other is A/A. Figure 26.2B shows the probability that this pattern would be observed, as a function of the recombination rate between the two genes, c—or in other words, the likelihood of the recombination rate, given these data. This is calculated using the simple rules of Mendelian inheritance. First, assume that A is linked with D, and a with d in the father (AD/ad). There are eight possible genotypes in the offspring: four different gametes can be inherited from the father, who is heterozygous at two loci, and two different gametes can be inherited from the mother, who is heterozygous at one locus. Adding up the possibilities, we find that the chances of an affected sibling with genotypes A/A, A/a are (1/4)c, (1/4) (1 – c), respectively, and for an unaffected sibling, (1/4) (2 – c), (1/4) (1 + c), respectively. The chance of seeing two affected siblings with A/a, one unaffected with A/A, and one unaffected with A/a, is therefore ((1/4)c) 2(1/4) (2 – c) (1/4) (1 + c). Now, it is equally likely that A is linked with d, and a with D in the father (Ad/ad)), in which case we replace c by (1 – c) in this formula. Averaging the two possible genotypes in the father, we have a likelihood
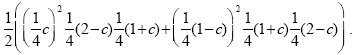
Because there is so little data—just one family—this does not give much information about the degree of linkage: It is only twice as likely that linkage is complete as that there is no linkage (Fig. 26.2B). However, multiplying likelihoods across many independent families can give accurate estimates of the recombination rate, c (Fig. 26.2C).
Figure 26.3 shows the total numbers of genes listed in the OMIM database over the past 40 years.
Comprehensive information about the human genome project is given at several websites: The National Human Genome Research Institute at the NIH, The Human Genome Project of the U.S. Department of Energy and the National Institutes of Health and The Human Genome at the Wellcome Trust.
Associations between Genetic Markers and Phenotype Can Tell Us the Genetic Basis of Complex Traits
Table 26.1 is from the Human Gene Mutation Database from Institute of Medical Genetics, Cardiff.
Figure 26.4 compares the statistical power of methods based on segregation within families with the transmission disequilibrium test (TDT), which is based on associations in the population as a whole. The TDT test was introduced by Spielman et al. (1993). This comparison was made by Risch and Merikangas (1996) in an influential paper that showed that genome-wide association studies should be possible with a feasible number of markers. We give details of the calculation .
Association Studies Are Based on Linkage Disequilibrium
The HapMap project is described here.
Zeggini et al. (2005) assess the power of tagging SNPs, using HapMap data.
African populations have higher genetic diversity, which implies longer coalescence times between genes and, hence, shorter regions with no recombination (p). Thus, there is a direct correspondence between higher diversity and lower linkage disequilibrium.
Searching the Genome for Associations with Quantitative Traits Is Difficult
The analysis of association studies for Alzheimer’s disease is from Emahazion et al. (2001).
Weiss and Terwilliger (2000), Terwilliger et al. (2002), and Terwilliger and Hiekkalinna (2006) take a pessimistic view of the statistical difficulties involved in genome-wide association studies.
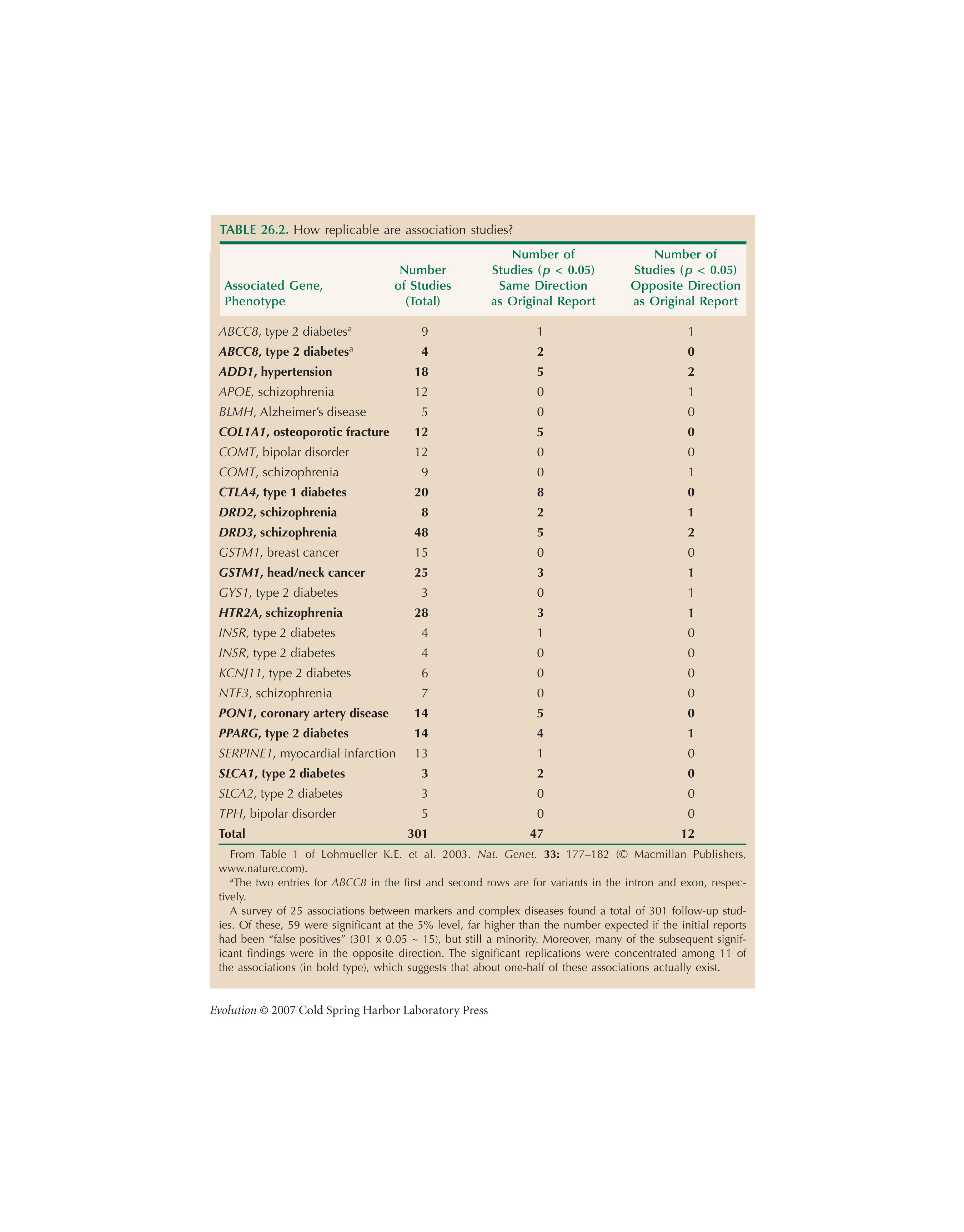
The meta-analysis of association studies in Table 26.2 is from Lohmueller et al. (2003).
A study by the Wellcome Trust Case-Control Consortium (2007) has recently published results from a genome-wide association study, which compares 2000 individuals affected by seven common diseases with 3000 controls. This large sample size allowed identification of 24 loci that are significantly associated with disease risk: 1 in bipolar disorder, 1 in coronary artery disease, 9 in Crohn’s disease, 3 in rheumatoid arthritis, 7 in type 1 diabetes, and 3 in type 2 diabetes. Most of these have been confirmed by replication. See also Sladek et al. (2007), who use HapMap data to identify an allele associated with susceptibility to diabetes.
Are There Common Variants for Common Diseases?
Corbo and Scacchi (1999) suggested that ApoE*4 is a “thrifty allele” that adapts to an ancestral nutrient-poor environment. Neel (1962) first introduced the idea of a “thrifty genotype” as an explanation for a high incidence of diabetes in the New World.
The importance of an allele for public health can be measured by its population attributable fraction (PAF), which can be thought of as the fraction of the disease that would be eliminated if the allele were not present. For example, in a Dutch population the ApoE*4 allele has a PAF of 20% for late-onset Alzheimer’s disease. This means that 20% of cases can be attributed to the presence of this allele (Carlson et al. 2004).
Mutations of SCN1A cause a variety of forms of epilepsy. See Graves (2006) for a review, and Nabbout et al. (2003) for the specific study of SMEI (severe myoclonic epilepsy in infancy).
What Can We Do with Genetic Information?
Many websites discuss the social implications of abundant genetic information, including the U.S. National Human Genome Research Institute (NHGRI), the Human Genome Organisation (HUGO), Genome Canada, The Nuffield Council on Bioethics, and The Wellcome Trust.
Implications for Medicine
There has been some extraordinary enthusiasm for the prospect of personalized medicine. For example, Weston and Hood (2004) expect that “[w]ithin the next 10–15 years, a predictive medicine will emerge, capable of determining a probabilistic, individualized future health history,” and Breithaupt (2001) says that “[t]he combination of genomics, genetic diagnosis technologies and powerful computers promises not less than to eradicate all leading causes of death in the First World that we have not yet found a cure for.” See also Breithaupt (2003). The Royal Society has published a more balanced report, which can be accessed on line here.
Screening policies for breast cancer are discussed at this NIH site and for Huntington’s disease are discussed here.
Prenatal Screening
Newberger (2000) reviews prenatal screening methods for Down syndrome.
For the possibility of isolating single cells from maternal blood or PAP smears, see Wang et al. (2004).
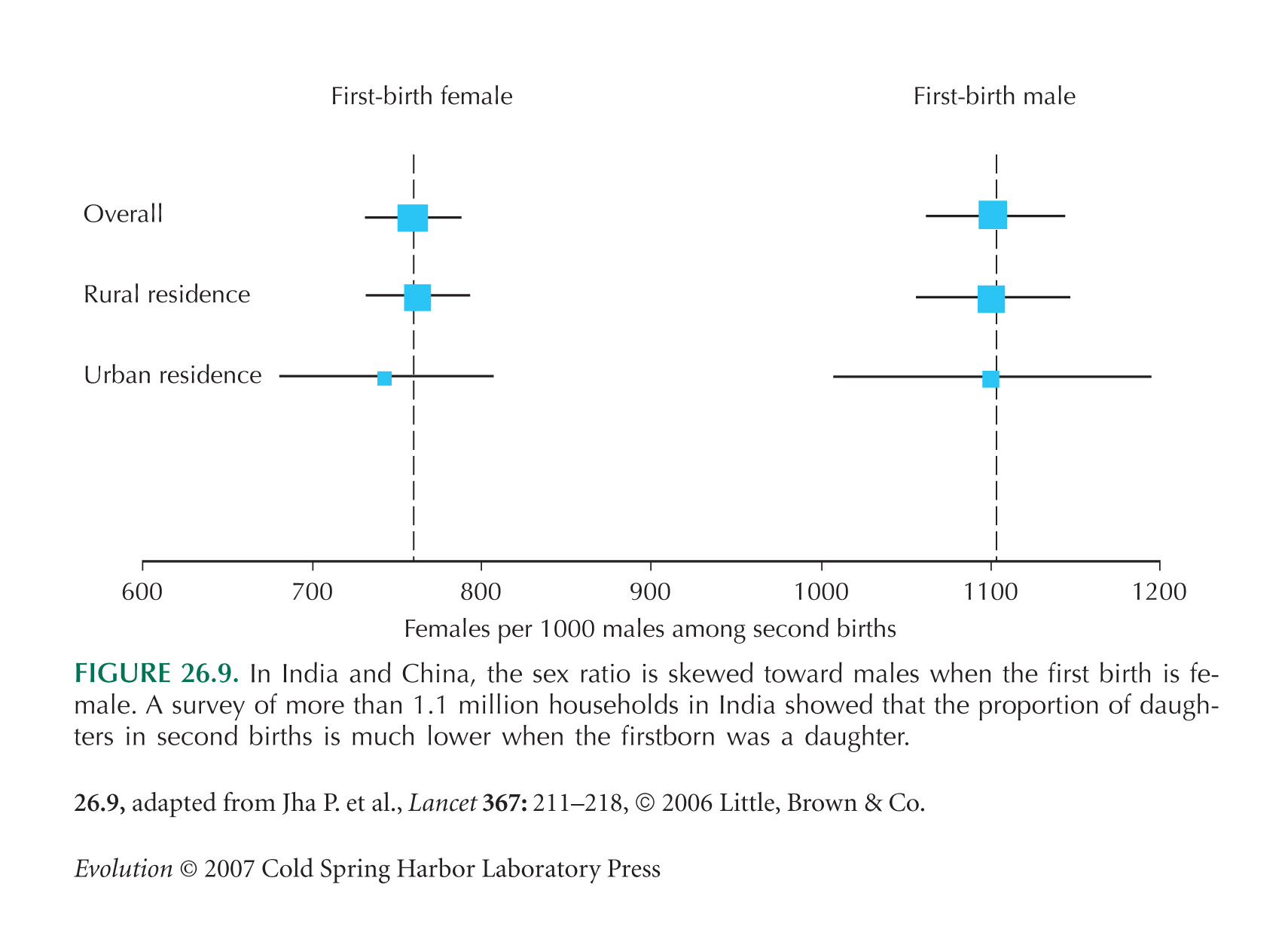
The sex ratio example of Figure 26.9 is from Jha et al. (2006).
Insurance
Two useful sources of information are the U.K. Forum on Genetics and Insurance and the U.S. National Conference of State Legislatures. See also Bonn (2000).
Identifying Individuals
The U.K. National DNA Database has an official website.
Ezzati et al. (2002) assess the contributions of major risk factors to disease burden. This is a useful example of current epidemiology.
Understanding Human Nature
Human Variation Cannot Be Subdivided into Discrete Races
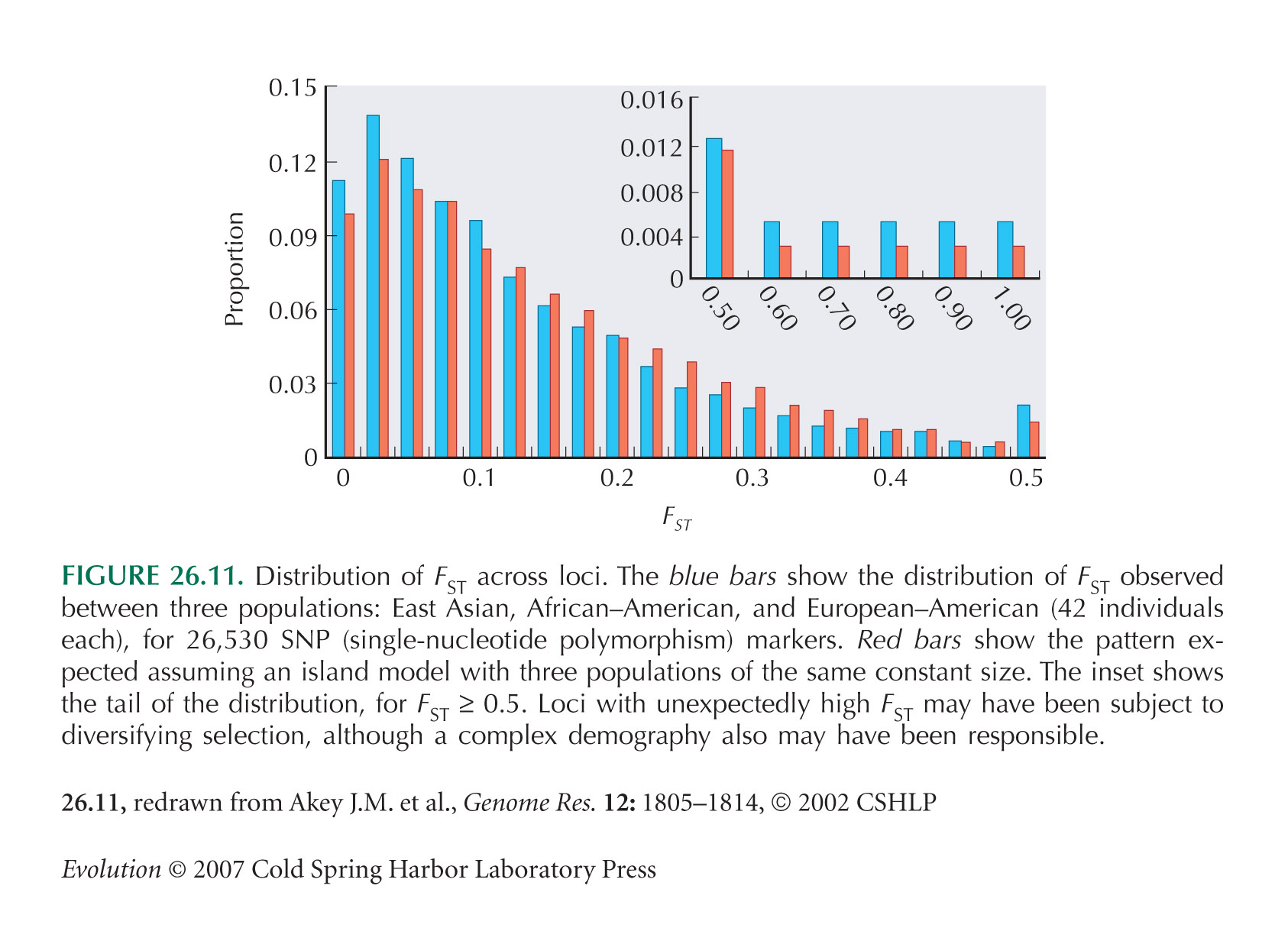
Akey et al. (2002) survey FST across large numbers of SNP markers (Fig. 26.11). See also Hinds et al. (2005, Suppl. Info.) and Kayser et al. (2003).
The analysis of genetic and linguistic boundaries across Europe is from Barbujani et al. (1990).
Tishkoff and Kidd (2004) and Tate and Goldstein (2004) discuss the implications of “racial” variation for medicine.
Natural Selection Has Shaped, and Is Shaping, Human Variation
McVean and Spencer (2006) give an excellent overview of how we can scan the human genome for evidence of selection. They make the point that genes which we already know to be selected (HLA, lactase, the Duffy locus, β-globin, and the 17q21 inversion) are detected in genome-wide scans. However, these are detected only by some measures, and not all, and these examples are at the extreme tails of the distribution of test statistics—suggesting that there are not many more cases that involve such strong selection. (They note that the CCR5 example, which is known to be under strong selection because it gives resistance to HIV infection, does not show a clear pattern in genome-wide scans.)
Voight et al. (2006) use data from the HapMap Project to scan the genome for signals indicating recent selective sweeps. They find a large number of candidates, although often restricted to one geographic region. Also see the Web Notes for Chapter 19.
Evidence for selection on lactose tolerance in Europe is given by Ennatah et al. (2002) and Bersaglier et al. (2004). Tishkoff et al. (2006) showed that lactose tolerance in Africa arose independently, but also through selection on the same gene. See Check (2006).
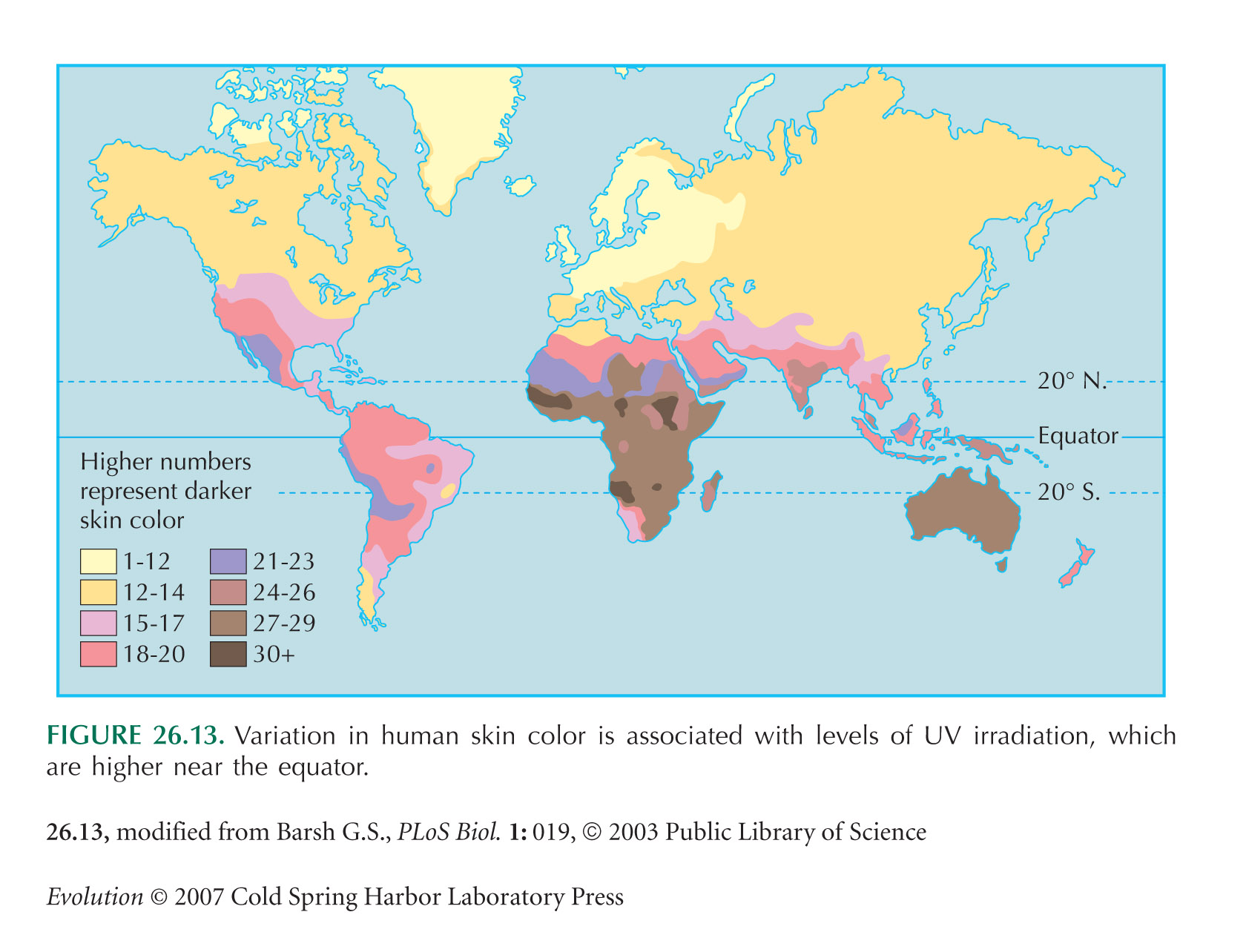
Diamond (2005) comments on Jablonski’s (2004) and Chaplin’s (2004) studies of the correlation between skin color and ultraviolet insolation. Barsh (2003) reviews the causes of human skin color variation.
The map in Figure 26.13 is from Barsh (2003). This raises the issue of whose skin color it measures: the Australian data are for the Aborigines, not European settlers, whereas the South African Cape gives the light skin color of the Boers. This might be justified on the grounds that European settlement of Australia is relatively recent (~200 years), whereas the Cape was settled by Boer and Bantu at around the same time, about 400 years ago. These kinds of decision are ultimately arbitrary, because tracing back our ancestry, we all came from elsewhere.
Harding et al. (2000) found that the candidate gene MCR1 had low diversity in Africa, suggesting a history of selection, but that this does not explain skin color variation. Recently, however, another gene has been found that does account for much skin color variation and that also shows evidence of recent selection (Lamason et al. 2005; Muller and Kelsh 2006).
A possible example of selection on genes that can alter human brain size has received considerable attention. We give details .
See Ponting and Jackson (2005) and McVean and Spencer (2006) for reviews.
Stefansson et al. (2005) show that a chromosomal inversion (17q21) is associated with figher female fecundity in the Icelandic population. This inversion is much more common in Europe than in Africa. However, it is ancient (around 1 Mya), and so other selective forces must prevent its fixation.
Hellmann et al. (2005) report a positive correlation between recombination rate and sequence diversity in humans, and argue that this is caused by selection at linked loci (see pp. 536–540 of Evolution)—another line of evidence for pervasive selection in the current human population. However, Spencer et al. (2006) argue that this correlation is more likely to be due to a direct mutagenic effect of recombination. At present, it is not clear to what extent selection at linked loci shapes neutral diversity in the human genome. See Web Notes for Chapter 23.
Nielsen et al. (2007) provide a recent review of population genetic evidence for selection on the human genome.
Relaxed Selection Will Eventually Lead to an Increased Incidence of Inherited Disease
Hamilton (1996) and Jones (2000) give opposing viewpoints on the consequences of relaxed selection for the future of our species.
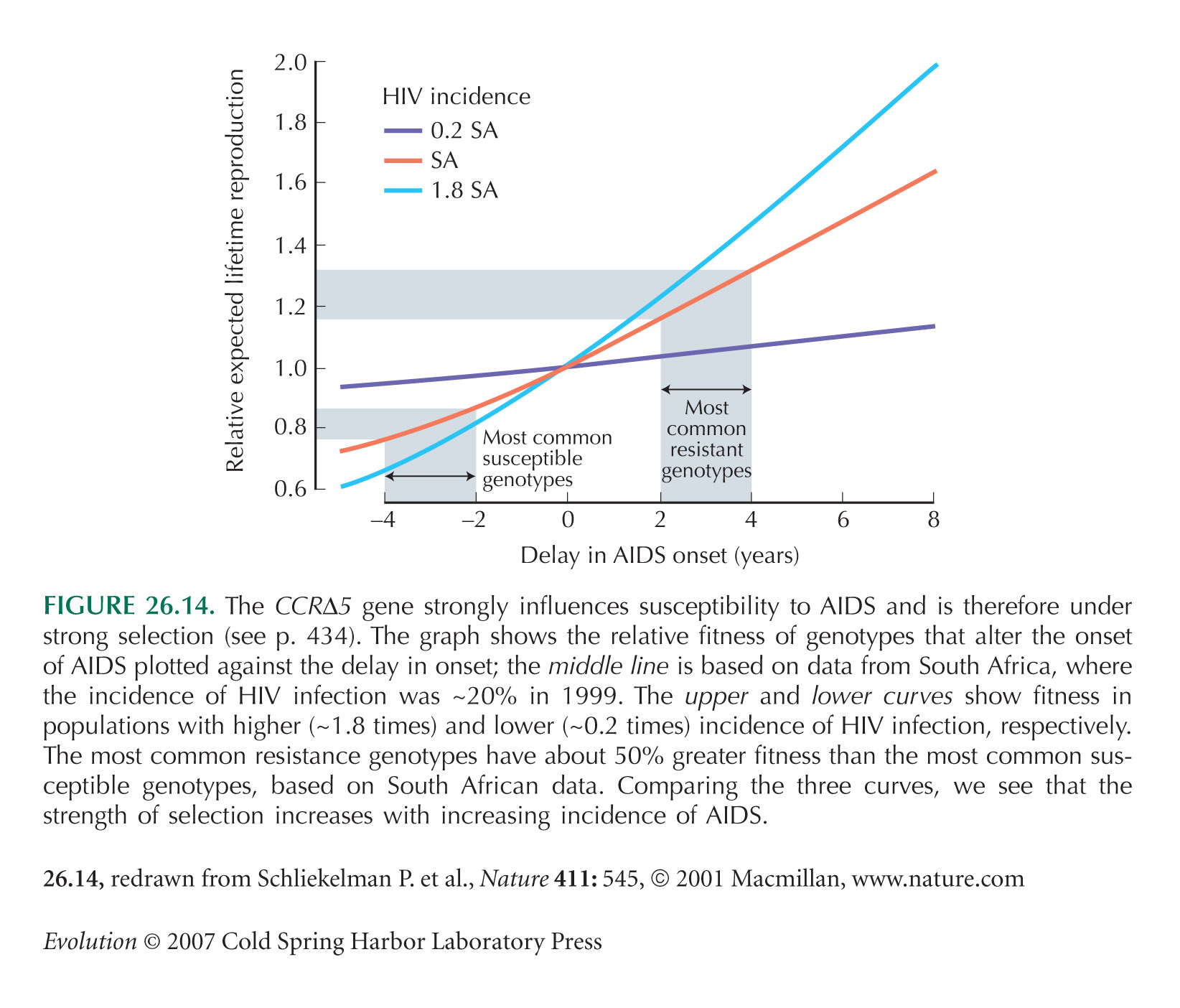
The CCRΔ5 example of Figure 26.14 is from Schliekelman et al. (2001).
Ferlin et al. (2006) review the genetic causes of male infertility, which include deletions on the Y chromosome.
Keightley et al. (2005, 2006) give interesting evidence that selection in the human lineage has been ineffective because our species has had a low effective population size. Keightley et al. (2005, 2006) show that there is no significant reduction in divergence between human and chimp in the 2-kb regions 5′ of genes, or 2-kb regions within intron 1. This contrasts with the mouse–rat comparison, where reduced divergence in these regions implies that ~17% of their sequence is constrained by selection. Keightley et al. (2005, 2006) argue that noncoding regions of hominid genomes accumulate mildly deleterious mutations because their effective population size is much lower than for rodents (Ne ~ 10–30,000 compared with 450,000–810,000 for Mus). They suggest that humans and chimpanzees have each accumulated ~140,000 slightly deleterious mutations, with selection ~10–4, since they diverged (see Box 18.1).
Bush and Lahn (2005, 2006) use a different method, which has higher resolution: They show that is substantially lower at sites adjacent to 16-bp windows that are conserved across human–mouse–dog or human–mouse–chicken. Thus, small regions are maintained by selection, but are not detected in Keightley et al.’s (2005, 2006) broader-scale comparison. However, the degree of constraint is substantially stronger when are compared, consistent with less effective selection in hominids.
Lynch and Conery (2003) and Lynch (2006) argue that species with small effective population size do lose functional sequences by random drift, but that this can lead to the evolution of new functions as other sites evolve to compensate for lost function. (See Web Notes for Chapter 24.)
Reed and Aquadro (2006) discuss the extent of selection that acts on our species and speculate on its consequences in the coming millennia. (However, note that their argument for pervasive selection rests partly on Hellmann et al. [2005], whose evidence has been questioned; see above.
Application of Genomic Medicine Depends on the Extent of Racial Variation
Branca (2005) comments on the controversy over licensing of BilDil specifically for African-Americans.
Ingelman-Sundberg (2005) reviews the pharmacological consequences of variation in CYP2D6, and discusses possible evolutionary explanations.
Darwinian Medicine Seeks Ultimate Explanations
The case for “Darwinian medicine” is made by Williams and Nesse (1991) and Nesse and Williams (1994). See also Lappe (1994) and Trevathan (1999).
Evolutionary Psychology Attempts to Understand Human Nature
Ruse (1979) and Kitcher (1985) review the early arguments over sociobiology, and Segerstrale (2000) takes a longer view.
There is a large literature on evolutionary psychology: see Further Reading (pp. 781–782) for some sources. Laland and Brown (2002) give a concise and critical explanation of the various approaches to understanding the evolution of human nature.
Darwin wrote almost nothing about our own species in On the Origin. However, near the end he wrote, “In the distant future I see open fields for far more important researches. Psychology will be based on a new foundation, that of the necessary acquirement of each mental power and capacity by gradation.” His Descent of Man (1871) and The Expression of Emotions in Man and Animals (1872) are the forerunners of sociobiology and evolutionary psychology.
Understanding How Human Nature Has Evolved Is Difficult
The example of nausea during pregnancy (Fig. 26.16) is from Flaxman and Sherman (2000).
Evolutionary explanations for human mate preferences are discussed by Buller (2005).
References
Akey J.M., Zhang G., Zhang K., Jin L., and Shriver M.D. 2002. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12: 1805–1814.
Barbujani G. and Sokal R.R. 1990. Zones of sharp genetic change in Europe are also linguistic boundaries. Proc. Natl. Acad. Sci. 87: 1816–1819.
Barsh G.S. 2003. What controls variation in human skin color? PLoS Biol. 1: e27.
Bell J. and Haldane J.B.S. 1937. The linkage between the genes for colour-blindness and haemophilia in man. Proc. R. Soc. (Lond.) B 123: 119–150.
Bernstein F. 1931. Zur Grundlegung der Vererbung beim Menschen. Z. Indukt. Abstammungs. Vererbungsl. 57: 113–138.
Bersaglier T., Sabeti P.C., Patterson N., Vanderploeg T., Schaffner S.F., Drake J.A., Rhodes M., Reich D.E., and Hirschhorn J.N. 2004. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 74: 1111–1120.
Bonn D. 2000. Genetic testing and insurance: Fears unfounded?. Lancet 355: 1526–1526.
Branca M.A. 2005. BiDil raises questions about race as a marker. Nat. Rev. Drug Discov. 4: 615–616.
Breithaupt H. 2001. The future of medicine. EMBO Rep. 2: 465–467.
Breithaupt H. 2003. Pioneers in medicine. EMBO Rep. 4: 1019–1021.
Buller D.J. 2005. Adapting minds. MIT Press, Cambridge, Massachusetts.
Bush E.C. and Lahn B.T. 2005. Selective constraint on noncoding regions of hominid genomes. PLoS Comput. Biol. 1: e73.
Bush E.C. and Lahn B.T. 2006. Understanding the degradation of hominid gene control: Author’s reply. PLoS Comput. Biol. 2: e26.
Carlson C.S., Eberle M.A., Kruglyak L., and Nickerson D.A. 2004. Mapping complex disease loci in whole-genome association studies. Nature 429: 446–452.
Chaplin G. 2004. Geographic distribution of environmental factors influencing human skin coloration. Am. J. Phys. Anthropol. 125: 292–302.
Check E. 2006. How Africa learned to love the cow. Nature 444: 994–996.
Corbo R.M. and Scacchi R. 1999. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a “thrifty” allele? Ann. Hum.Genet. 63: 301–310.
Crow J. 1993. Felix Bernstein and the first human marker locus. Genetics 133: 4–7.
Currat M., Excoffier L., Maddison W., Otto S.P., Ray N., Whitlock M.C., and Yeaman S. 2006. Comment on “Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens” and “Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans.” Science 313: 172.
Darwin C. 1871. The descent of man, and selection in relation to sex. John Murray, London.
Darwin C. 1872. The expression of emotions in man and animals. John Murray, London.
Dediu D. and Ladd D.R. 2007. Linguistic tone is related to the population frequency of the adaptive haplogroups of two brain size genes, ASPM and Microcephalin. Proc. Natl. Acad. Sci. 104: 10944–10949.
Diamond J. 2005. Geography and skin colour. Nature 435: 283–284.
Emahazion T., Feuk L., Jobs M., Sawyer S.L., Fredman D., St Clair D., Prince J.A., and Brookes A.J. 2001. SNP association studies in Alzheimer’s disease highlight problems for complex disease analysis. Trends Genet. 17: 407–413.
Ennatah N.S., Sahi T., Savilahti E., Terwilliger J.D., Peltonen L., and Jarvela I. 2002. Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 30: 233–237.
Evans P.D., Anderson J.R., Vallender E.J., Choi S.S., and Lahn B.T. 2004. Reconstructing the evolutionary history of Microcephalin, a gene controlling human brain size. Hum. Mol. Genet. 13: 1139–1145.
Evans P.D., Gilbert S.L., Mekel-Bobrov N., Vallender E.J., Anderson J.R., Vaez-Azizi L.M., Tishkoff S.A., Hudson R.R.,and Lahn B.T. 2005. Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 309: 1717–1720.
Ezzati M., Lopez A.D., Rodgers A., Vander Hoorn S., Murray C.J.L., and C.R.A.C. Group. 2002. Selected major risk factors and global and regional burden of disease. Lancet (online)
Ferlin A., Arredi B., and Foresta C. 2006. Genetic causes of male infertility. Reprod. Toxicol. 22: 133–141.
Flaxman S.M. and Sherman P.W. 2000. Morning sickness: A mechanism for protecting mother and embryo. Q. Rev. Biol. 75: 113–148.
Graves T.D. 2006. Ion channels and epilepsy. QJM 99: 201–217.
Haldane J.B.S. and Smith C.A.B. 1947. A new estimate of the linkage between the genes for haemophilia and colour-blindness in man. Ann. Eugen. 14: 10–31.
Hamilton W.D. 1996. Narrow roads of gene land, Volume 1: Evolution of social behaviour. W.H. Freeman, Oxford.
Harding R.M., Healy E., Ray A.J., Ellis N.S., Flanagan N., Todd C., Dixon C., Sajantila A., Jackson I.J., Birch-Machin M.A., and Rees J.L. 2000. Evidence for variable selective pressures at MC1R. Am. J. Hum. Genet. 66: 1351–1361.
Hellmann I., Prüfer K., Ji H., Zody M.C., Pääbo S., and Ptak S.E. 2005. Why do human diversity levels vary at a megabase scale? Genome Res. 15: 1222–1231.
Hinds D.A., Stuve L.L., Nilsen G.B., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., and Cox D.R. 2005. Whole-genome patterns of common DNA variation in three human populations. Science 307: 1072–1079.
Ingelman-Sundberg M. 2005. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 5: 6–13.
Jablonski N.G. 2004. The evolution of human skin and skin color. Annu. Rev. Anthropol. 33: 585–623.
Jha P., Kumar R., Vasa P., Dhingra N., Thiruchelvam D., and Moineddin R. 2006. Low male-to-female sex ratio of children born in India: National survey of 1.1 million households. Lancet 367: 211–218.
Jones J.S. 2000. The language of the genes. Flamingo, London.
Kayser M., Brauer S., and Stoneking M. 2003. A genome scan to detect candidate regions influenced by local natural selection in human populations. Mol. Biol. Evol. 20: 893–900.
Keightley P.D., Lercher M.J., and Eyre-Walker A. 2005. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 3: e42.
Keightley P.D., Lercher M.J., and Eyre-Walker A. 2006. Understanding the degradation of hominid gene control. PLoS Comput. Biol. 2: e19.
Kitcher P. 1985. Vaulting ambition: Sociobiology and the quest for human nature. MIT Press, Cambridge, Massachusetts.
Kouprina N., Pavlicek A., Mochida G.H., Solomon G., Gersch W., Yoon Y.H., Collura R., Ruvolo M., Barrett J.C., Woods C.G., et al. 2004. Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biol. 2: E126.
Laland K. and Brown G.G. 2002. Sense and nonsense: Evolutionary perspectives on human behaviour. Oxford University Press, Oxford.
Lamason R.L., Mohideen M.A., Mest J.R., Wong A.C., Norton H.L., Aros M.C., Jurynec M.J., Mao X., Humphreville V.R., Humbert J.E., et al. 2005. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310: 1782–1786.
Lappe M. 1994. Evolutionary medicine: Rethinking the origins of disease. Random House, New York.
Lohmueller K.E., Pearce C.L., Pike M.C., Lander E.S., and Hirschorn J.N. 2003. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 33: 177–182.
Lynch M. 2006. The origins of eukaryotic gene structure. Mol. Biol. Evol. 23: 450–468.
Lynch M. and Conery J.S. 2003. The origins of genome complexity. Science 302: 1401–1404.
McVean G. and Spencer C.C.A. 2006. Scanning the human genome for signals of selection. Curr. Opin. Genet. Dev. 16: 624–629.
Mekel-Bobrov N., Evans P.D., Gilbert S.L., Vallender E.J., Hudson R.R., and Lahn B.T. 2006. Response to “Comment on ‘Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens’ and ‘Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans.’” Science 313: 172.
Mekel-Bobrov N., Gilbert S. L., Evans P.D., Vallender E.J., Anderson J.R., Hudson R.R., Tishkoff S.A.,and Lahn B.T. 2005. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 309: 1720–1722.
Mekel-Bobrov N., Posthuma D., Gilbert S.L, Lind P., Gosso M.F., Luciano M., Harri S.E., Bates T.C., Polderman T.J.C., Whalley L.J., et al. 2007. The ongoing adaptive evolution of ASPM and Microcephalin is not explained by increased intelligence. Hum. Mol. Genet. 16: 600–608.
Morton N.E. 1995. LODs past and present. Genetics 140: 7–12.
Muller J. and Kelsh R.N. 2006. A golden clue to human skin colour variation. BioEssays 28: 578–582.
Nabbout R., Gennaro E., Dalla Bernardina B., Dulac O., Madia F., Bertini E., Capovilla G., Chiron C., Cristofori G., Elia M., et al. 2003. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology 60: 1961–1967.
Neel J.V. 1962. Diabetes mellitus: A “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 14: 353–362.
Nesse R.M. and Williams G.C. 1994. Why we get sick: The new science of Darwinian medicine. Crown Publishing Group, New York.
Newberger D.S. 2000. Down Syndrome: Prenatal risk assessment and diagnosis. Am. Fam. Physician 62: 825–832.
Nielsen R., Hellmann I., Hubisz M., Bustamante C., and Clark A.G. 2007. Recent and ongoing selection in the human genome. Nat. Rev. Genet. 8: 857–868.
O’Rahilly S., I. Barroso, and Wareham N.J. 2005. Genetic factors in type 2 diabetes: The end of the beginning? Science 307: 370–373.
Ott J. 1999. Analysis of human genetic linkage. Johns Hopkins University Press, Baltimore.
Ponting C.P. and Jackson A.P. 2005. Evolution of primary microcephaly genes and the enlargement of primate brains. Curr. Opin. Genet. Dev. 15: 241–248.
Reed F.A. and Aquadro C.F. 2006. Mutation, selection and the future of human evolution. Trends Genet. 22: 479–484.
Risch N. and Merikangas K. 1996. The future of genetic studies of complex human diseases. Science 273: 1516–1517.
Ruse M. 1979. Sociobiology: Sense or nonsense? Reidel, Dordrecht/Boston.
Rushton J.P., Vernon P.A., and Bons T.A. 2007. No evidence that polymorphisms of brain regulator genes Microcephalin and ASPM are associated with general mental ability, head circumference or altruism. Biol. Lett. 3: 157–160.
Schliekelman P., Garner C., and Slatkin M. 2001. Natural selection and resistance to HIV. Nature 411: 545–545.
Segerstrale U. 2000. Defenders of the truth: The battle for science in the sociobiology debate and beyond. Oxford University Press, Oxford.
Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S., et al. 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885.
Spencer C.C.A., Deloukas P., Hunt S., Mullikin J.C., Myers S., Silverman B., Donnelly P., Bentley D., and McVean G. 2006. The influence of recombination on human genetic diversity. PLoS Genet. 2: e148.
Spielman R.S., McGinnis R.E., and Ewens W.J. 1993. Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52: 506.
Stefansson H., Helgason A., Thorleifsson G., Steinthorsdottir V., Masson G., Barnard J., Baker A., Jonasdottir A.I.A., Gudnadottir V.G., Desnica N., et al. 2005. A common inversion under selection in Europeans. Nat. Genet. 37: 129–137.
Tate S.K. and Goldstein D.B. 2004. Will tomorrow’s medicines work for everyone? Nat. Genet. 36: S34–S42.
Terwilliger J.D. and Hiekkalinna T. S. 2006. An utter refutation of the “Theorem of the HapMap.” Eur. J. Hum. Genet. 14: 426–437.
Terwilliger J.D. and Ott J. 1994. Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore.
Terwilliger J.D., Haghighi F., Hiekkalinna T.S., and Goring H.H. 2002. A bias-ed assessment of the use of SNPs in human complex traits. Curr. Opin. Genet. Dev. 12: 726–734.
Tishkoff S.A. and Kidd K.K. 2004. Implications of biogeography of human populations for “race” and medicine. Nat. Genet. 36: S21–S27.
Tishkoff S.A., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., et al. 2006. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 39: 31–40.
Trevathan W.R. 1999. Evolutionary medicine. Oxford University Press, Oxford.
Vogel F. and Motulsky A.G. 1997. Human genetics: Problems and approaches. Springer-Verlag, Berlin.
Voight B.F., Kudaravalli S., Wen X., and Pritchard J.K. 2006. A map of recent positive selection in the human genome. PLoS Biol. 4: e72.
Wang T., Chen H., and Ma T. 2004. Noninvasive prenatal diagnosis of fetal sex by single-cell PEP-PCR method. J. Huazhong Univ. Sci. Technolog. Med. Sci. 24: 66–78.
Wang Y.Q. and Su B. 2004. Molecular evolution of Microcephalin, a gene determining human brain size. Hum. Mol. Genet. 13: 1131–1137.
Weiss K.M. and Terwilliger J.D. 2000. How many diseases does it take to map a gene with SNPs? Nat. Genet. 26: 151–157.
Wellcome Trust Case-Control Consortium. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–668.
Weston A.D. and Hood L. 2004. Systems biology, proteomics, and the future of health care: Toward predictive, preventative, and personalized medicine. J. Proteome Res. 3: 179–196.
Williams G.C. and Nesse R.M. 1991. The dawn of Darwinian medicine. Q. Rev. Biol. 66: 1–22.
Woods R.P., Freimer N.B., De Young J.A., Fears S.C., Sicotte N.L., Service S.K., Valentino D.J., Toga A.W., and Mazziotta J.C.. 2006. Normal variants of Microcephalin and ASPM do not account for brain size variability. Hum. Mol. Genet. 15: 2025–2029.
Zeggini E., Rayner W., Morris A.P., Hattersley A.T., Walker M., Hitman G.A., Deloukas P., Cardon L.R., and McCarthy M.I. 2005. An evaluation of HapMap sample size and tagging SNP performance in large-scale empirical and simulated data sets. Nat. Genet. 37: 1320–1322.
Zhang J. 2003. Evolution of the human ASPM gene, a major determinant of brain size. Genetics 165: 2063–2070.
WWW References
http://cancernet.nci.nih.gov/cancertopics/pdq/genetics/breast-and-ovarian/healthprofessional Genetics of Breast and Ovarian Cancer (PDQ<r>). National Cancer Institute (NCI), U.S. National Institutes of Health (NIH), Bethesda, Maryland.
http://genome.wellcome.ac.uk/geneticsandsociety/ Genetics and Society. Wellcome Trust, London.
http://genome.wellcome.ac.uk/node30120.html The Human Genome. The Wellcome Trust. London.
http://www.genome.gov/10001772 National Human Genome Research Institute (NHGRI), Bethesda, Maryland.
http://www.genome.gov/PolicyEthics/ Policy & Ethics, critical issues and legislation surrounding genetic issues. National Human Genome Research Institute (NHGRI), Bethesda, Maryland.
http://www.genomecanada.ca/en/info/ethical/ Ethical, Environmental, Economic, Legal and Social Implications of Genomic Research. Genome Canada, Ottawa.
http://www.hapmap.org/whatishapmap.html The International HapMap Project.
http://www.hdsa.org Huntington’s Disease Society of America.
http://www.hgc.gov.uk/Client/Content_wide.asp?ContentId=470 UK Forum for Genetics and Insurance. Human Genetics Commission (HGC), London.
http://www.hgmd.cf.ac.uk The Human Gene Mutation Database. Institute of Medical Genetics, Cardiff, United Kingdom.
http://www.homeoffice.gov.uk/science-research/using-science/dna-database/ The national DNA database. Home Office, London.
http://www.hugo-international.org/comm_hugoethicscommittee.php HUGO Ethics Committee. Human Genome Organisation, Singapore.
http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM Online Mendelian Inheritance in Man®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland.
http://www.ncsl.org/programs/health/genetics/ndislife.htm Genetics and Life, Disability and Long-term Care Insurance. National Conference of State Legislatures, Denver, Colorado and Washington, D.C.
http://www.nuffieldbioethics.org Nuffield Council on Bioethics, London.
http://www.ornl.gov/sci/techresources/Human_Genome/home.shtml The Human Genome Project. Site sponsored by the U.S. Department of Energy Office of Science, Office of Biological and Environmental Research, Human Genome Program, the U.S. Department of Energy.
http://www.royalsoc.ac.uk/document.asp?tip=0&id=3780 Personalised medicines: Hopes and realities. 21 September 2005. The Royal Society, London.
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}